

Fenilketonurija (PKU) je autosomno recesivno nasledna metabolopatija, uslovljena mutacijom strukturnog gena za sintezu jetrinog enzima fenilalanin-hidroksilaze i/ili kofaktora tetrahidrobiopterina. Fenilalanin se ne razgrađuje u organizmu, nego se nagomilava, uzrokujući oštećenja centralnog nervnog sistema. Problem je u tome što se ova aminokiselina nalazi u gotovo svim namirnicama.

Klinička slika fenilketonurije
Novorođenčad su asimptomna, ali se ubrzo javlja uporno povraćanje. Ukoliko se ne leči, prevelika količina fenilalanina u krvotoku dovodi do oštećenja mozga kod dece, što je uzrok mentalne retardacije i zaostalosti u razvoju (deca ne drže glavu, ne sede). Nedovoljna količina tirozina u krvotoku vodi do smanjenja produkcije pigmenta melanin, tako da deca sa ovim poremećajem su pretežno bleda, imaju nežno belu boju kože i plave oči. Neretko su svetlije puti od članova porodice.
Usled nedostatka enzima, fenilalanin se prebacuje u oblik fenilketona, koji se izbacuje u mokraću, tj. urin. Usled ovih ketona, znoj i urin obolelih imaju intenzivniji miris od zdravih osoba - miris na vlagu.
Postavljanje dijagnoze fenilketonurije
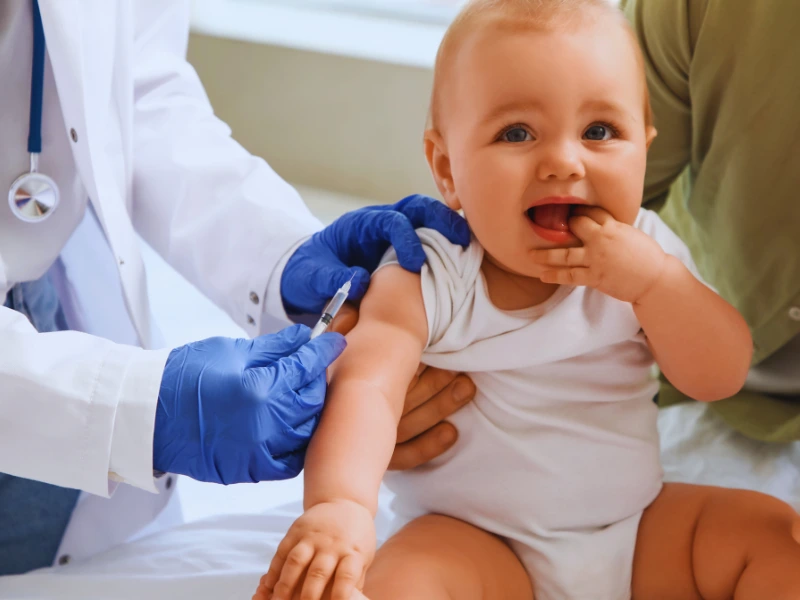
Svakom novorođenom detetu na teritoriji uže Srbije i Vojvodine, kao i u većini zemalja sveta, u trećem danu života uzima se uzorak krvi za analizu - Gutrijev test. Ako se ustanovi da su koncentracije fenilalanina povišene, deca se pozivaju na dopunsko ispitivanje.
Trudnoća i fenilketonurija
Za žene sa fenilketonurijom (PKU) koje planiraju potomstvo, adekvatna terapija nije samo pitanje ličnog zdravlja, već jedan od najvažnijih faktora za pravilan razvoj ploda. Nelečena fenilketonurija tokom trudnoće nosi visok rizik od spontanog pobačaja ili zastoja u razvoju fetusa. Deca majki sa visokim nivoom fenilalanina u krvi mogu se suočiti sa ozbiljnim zdravstvenim izazovima, uključujući mikrocefaliju (abnormalno malu glavu), urođene srčane mane, kao i različite razvojne promene.
S obzirom na to da su ove komplikacije direktno povezane sa nivoom ove aminokiseline u krvi majke, imperativ je da se svaka žena koja je ranije prekinula lečenje vrati strogo kontrolisanom režimu ishrane i terapiji pre samog začeća. Tokom čitave trudnoće neophodan je kontinuiran nadzor stručnog tima, koji prvenstveno čine genetičar i dijetetičar, kako bi se nivoi fenilalanina održali u bezbednim okvirima za razvoj bebe.
Lečenje fenilketonurije
Lečenje se sprovodi već od 3. nedelje života, a tada se započinje ishranom specijalnim preparatima koji ne sadrže fenilalanin. Minimalne, neophodne količine fenilalanina obezbeđuju se iz mleka, a kasnije i iz drugih namirnica. Dijeta se organizuje isključivo uz lekarsku kontrolu i sprovodi ceo život. Uspeh terapije je dobar, IQ je normalan, ali su neki manji poremećaji mogući, kao smetnje u pisanju, govoru i socijalnom ponašanju.
Lekari kod kojih možete zakazati pregled:
- Prim. dr Jelena Tomić, pedijatar i imunolog,
- Prim. dr Vesna Veličković, pedijatar i imunolog,
- Spec. dr med. Gordana Petrović, pedijatar i imunolog,
- Spec. dr Nevenka Raketić, pedijatar i imunolog.
- Mr sci. med. dr Mirjana Dukić Jovanović, imunolog,
- Spec. dr med. Žikica Jovičić, imunolog,
- Prof. dr Sanvila Rašković, imunolog,
- Prof. dr Anđelka Stojković, pulmolog,
- Prof. dr Tatjana Pejčić, pulmolog,
- Prim. dr sci. med. Tatjana Radosavljević, pneumoftiziolog.
Povezani tekstovi:



Broj komentara: 3
Prijavite se
Dobro došli! Unesite svoje login podatke
Poštovanje, da li neko može da mi odgovori da li se pojava fenilketonurije može isključiti prenatalnim testom, kojim su analizirani svi hromozomi? Hvala
Vaš komentar nam je veoma dragocen, molimo upišite ga ovde
Porodica i zdravlje